GRASP command-line interface (CLI)
There is a command-line interface of GRASP that can prove useful if you want to automate tasks, run reconstructions on your own dedicated hardware, and/or access the latest features. This version is essentially a command-line interface to the backend features of the web-based service. It is worth noting that the web-based version has the advantage of a visual user interface, but that also means that it may lack the latest functionality.
The command-line version allows access to a variety of indel inference approaches. Beyond the default bi-directional edge encoding (BE), indels are available by either Position Specific (PS), or Simple Indel Coding (SIC). Regardless of encoding, indels can be inferred using either Parsimony (P) or Maximum Likelihood (ML) methods. This gives six methods: PS-P, PS-ML, SIC-P, SIC-ML, BE-P, BE-ML.
The command-line version accepts a file with evolutionary rates inferred with the tree, as produced by several tools incl. IQ-TREE2. At the moment the web-based version does not.
The command-line interface is implemented in bnkit as a class asr.GRASP.
asr.GRASP: What can it do?
asr.GRASP accepts an alignment (FASTA or Clustal formats) and a phylogenetic tree (Newick format) with concordant labels, to infer ancestor sequences by joint or marginal reconstruction by maximum likelihood. In the process, the program also infers insertion and deletion events, which are internally represented via partial-order graphs; it also identifies the most supported path of sequence inclusions at each ancestor.
The program can save all ancestor sequences (in the case of joint reconstruction) or one sequence (in the case of marginal reconstrution; optionally with character state distributions as a TSV file). It can save the partial-order graphs in JSON or as DOT files, which can be visualised with GraphViz. It can also re-save the tree with assigned ancestor labels.
GRASP was designed primarily for protein sequences but the command-line version incorporates DNA models too. At this stage we have not tested DNA sequence functionality extensively, nor have we developed specific features around DNA sequences (codon-centric analyses, user-provided background stats, etc).
asr.GRASP: How do I make it work on my computer?
First, you will need Java version 8 or newer. Any operating system with Java should work, including Mac OS, MS Windows and Linux.
Then, you have a choice: you can clone/download bnkit in its entirety. You may need JUnit 5 testing to get everything working; this is only required if you want to run software tests, say if you are a developer.
Alternatively, just download the pre-compiled version with all indel inference methods bnkit JAR file. This is the 21st of March 2024 version and compiled with a recent version of Java (19).
We also offer a version compiled using Java 11.
Or, the legacy version bnkit JAR file, which we keep to ensure reproducibility of results presented in the original papers.
asr.GRASP: How do I run it?
-
Download the jar file
We suggest that you then follow steps 2 onwards, but likely you can simply run it from the directory to which it was downloaded, e.g.
java -jar ~/Downloads/bnkit.jarshould produce the help info below. -
Create a bash script grasp that contains the following two lines, replacing the path with the path to your downloaded jar
#!/bin/sh java -jar -Xmx16g </path/to/bnkit.jar> $@(the
-Xmxis optional; see below) -
Change permissions on the bash script
chmod 755 grasp
- Place the file
graspwhere you store your executable files, for example/usr/local/bin
mv grasp /usr/local/bin
- Check that it works
grasp -h
This will print out the arguments that specifies your input data and options.
A typical command may look like this
grasp -aln 500_2112_dhad_18032019.aln -nwk r_500_2112_dhad_18032019.nwk -out recon_0500.aln -verbose -gap -thr 5
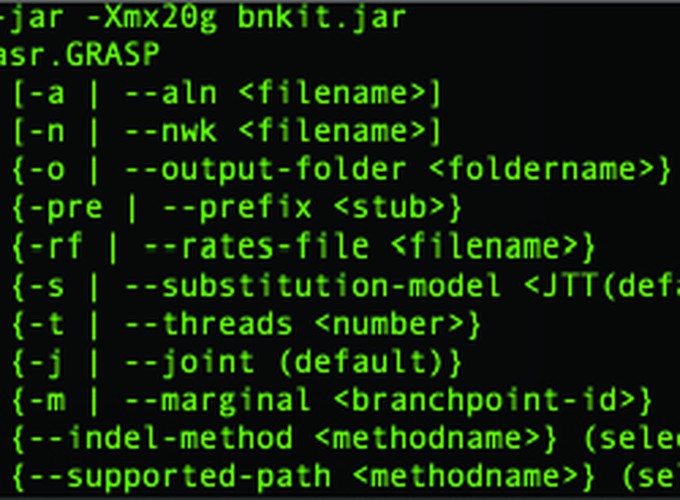
Full help information
Usage: asr.GRASP
[-a | --aln <filename>]
[-n | --nwk <filename>]
{-o | --output-folder <foldername>} (default is current working folder, or input folder if available)
{-i | --input-folder <foldername>}
{-pre | --prefix <stub>}
{-rf | --rates-file <filename>}
{-s | --substitution-model <JTT(default)|Dayhoff|LG|WAG|JC|Yang>}
{-t | --threads <number>}
{-j | --joint (default)}
{-m | --marginal <branchpoint-id>}
{--indel-method <methodname>} (select one from BEP(default) BEML SICP SICML PSP PSML)
{--supported-path <methodname>} (select one from DIJKSTRA(default) ASTAR)
{--nogap}
{--nonibble}
{--exclude-noedge}
{--save-as <list-of-formats>} (select multiple from FASTA CLUSTAL TREE DISTRIB ASR DOT TREES)
{--save-all} (saves reconstruction with ALL formats)
{--save-tree} (bypasses inference and re-saves the tree with ancestor nodes labelled as per GRASP's
depth-first labelling scheme starting with N0)
{--save-poag { <branchpoint-id> } (bypasses inference and saves the input alignment as a POAG
(partial order alignment graph of extant sequences under specified ancestor [default N0])
{--time}{--verbose}{--help}
Inference is a two-stage process:
(1) A history of indel events is inferred by either maximum likelihood or maximum parsimony and
mapped onto the tree to determine what positions contain actual sequence content
(2) For each ancestral position, the most probable character is assigned to each phylogenetic branch
point when performing a joint reconstruction. Alternatively, for each
position at a nominated branch point, the probability distribution over all possible
characters is inferred when performing a marginal reconstruction.
Finally, edges are drawn to represent all inferred combinations of indels to form an ancestor POG
with nodes that can form a valid sequence with inferred content; a preferred path
through the POG is then inferred, nominating a single, best supported sequence.
Mode of character inference:
-j (or --joint) activates joint reconstruction (default),
-m (or --marginal) activates marginal reconstruction (requires a branch-point to be nominated)
--onlyindel disengages the stage of character state inference
Required arguments:
-a (or --aln) must specify the name of a multiple-sequence alignment file on FASTA or CLUSTAL format
-n (or --nwk) must specify the name of a phylogenetic-tree file on Newick format
Optional arguments:
-o (or --output-folder) specifies the folder that will be used to save output files,
e.g. inferred ancestor or ancestors, tree, etc. as specified by format
-i (or --input-folder) skips indel inference, and loads a previous reconstruction from specified folder
-sa (or --save-as) lists the files and formats to be generated (see below)
--save-all nominates all
-pre (or --prefix) specifies a stub that is added to result filenames (default is the prefix of the alignment file)
-indel (or --indel-method) specifies what method to use for inferring indels (see below)
-s (or --substitution-model) specifies what evolutionary model to use for inferring character states (see below)
-rf (or --rates-file) specifies a tabulated file with relative, position-specific rates
We recommend the use of this generally, but specifically for trees with great distances, and with biologically diverse entries
As an example, IQ-TREE produces rates on the accepted format
--include-extants means that extants are included in output files (when the format allows)
--nogap means that the gap-character is excluded in the resulting output (when the format allows)
--nonibble de-activates the removal of indices in partial order graphs that cannot form a path from start to end
--orphans de-activates the removal of orphaned indel trees
--exclude-noedge removes non-existing edge as an option for parsimony in BEP
--verbose prints out information about steps undertaken, and --time the time it took to finish
-h (or --help) will print out this screen
Files/formats:
FASTA: sequences (most preferred path at each ancestor, gapped or not gapped)
CLUSTAL: sequences (most preferred path at each ancestor, gapped)
TREE: phylogenetic tree with ancestor nodes labelled
DISTRIB: character distributions for each position (indexed by POG, only available for marginal reconstruction)
ASR: complete reconstruction as JSON, incl. POGs of ancestors and extants, and tree (ASR.json)
DOT: partial-order graphs of ancestors in DOT format
TREES: position-specific trees with ancestor states labelled
Indel-methods:
BEP: bi-directional edge (maximum) parsimony
BEML: bi-directional edge maximum likelihood (uses uniform evolutionary model akin to JC)
SICP: simple indel-coding (maximum) parsimony (based on Simmons and Ochoterena)
SICML: simple indel-coding maximum likelihood (uses uniform evolutionary model)
PSP: position-specific (maximum) parsimony
PSML: position-specific maximum likelihood (uses uniform evolutionary model)
Add '*' to method name for less conservative setting (if available)
Substitution-models:
JTT: Jones-Taylor-Thornton (protein; default)
Dayhoff: Dayhoff-Schwartz-Orcutt (protein)
LG: Le-Gasquel (protein)
WAG: Whelan-Goldman (protein)
JC: Jukes-Cantor (DNA)
Yang: Yang's general reversible process model (DNA)
Notes:
Greater number of threads may improve processing time up to a point when coordination chokes performance; default is 4 threads.
Running GRASP requires large memory and in most cases Java needs to be run with the option -Xmx20g,
where 20g specifies that 20GB of RAM should be available.
~ This is version 21-Mar-2024 ~
Access through Docker
GRASP is available through Docker Hub at gabefoley/grasp
Once you have docker installed you can
docker run -it -v {full path to where your data is located}:/data gabefoley/grasp grasp -aln /data/{name of your alignment file}.aln -nwk /data/{name of your newick file}.nwk -out /data
for example, for me the command looks like (I have test_6.aln and test_6.nwk sitting in a /data folder):
docker run -it -v /Users/coolusername/Documents/code/grasp/data:/data grasp-docker grasp -aln /data/test_6.aln -nwk /data/test_6.nwk -out /data
This should give you a file, GRASP_ancestors.fasta appearing in folder: /Users/coolusername/Documents/code/grasp/data.
What else?
Running the command-line version is typically a quicker affair, at least for smaller reconstructions, but it requires decent hardware. A reconstruction of less than 1,000 sequences should take less than 10 minutes.
You can probably run a reconstruction with 10,000 sequences on a server, but how “gappy” the alignment is will also play a part in deciding this. If the alignment is reasonably clean, a powerful, modern laptop with at least 16GB of memory, can do this in under a day. If the alignment covers a diverse family, you will probably need a lot more memory. We recommend you set the Java heap size to 60GB RAM, which you can using the option -Xmx60000m.
The rough estimates above assume you use multiple threads; we recommend 5 or so on decent hardware (--threads 5).